
En el vasto y silencioso universo que habita dentro de cada una de nuestras células, se libra una batalla constante de creación y orden. Es un cosmos en miniatura donde miles de millones de nanomáquinas moleculares, conocidas como proteínas, ejecutan una sinfonía incesante que sustenta la vida misma. Estas máquinas no son de metal ni de silicio; son cadenas exquisitamente largas de moléculas más pequeñas, los aminoácidos, que, tras ser ensambladas, deben realizar un acto de origami molecular casi milagroso: plegarse sobre sí mismas para adoptar una estructura tridimensional única y precisa.
Esta forma tridimensional no es un capricho estético de la naturaleza; es el origen de su función. Una proteína correctamente plegada puede transportar oxígeno, digerir alimentos, transmitir señales nerviosas o combatir infecciones. Una proteína mal plegada, sin embargo, puede convertirse en un agente de caos, un saboteador molecular que desencadena enfermedades devastadoras como el alzhéimer, el párkinson o la fibrosis quística.
Durante medio siglo, predecir la forma final de una proteína a partir de su secuencia lineal de aminoácidos fue uno de los mayores desafíos de la biología, un enigma tan complejo que parecía casi irresoluble. En 2020, el mundo científico contuvo la respiración cuando AlphaFold2, un sistema de inteligencia artificial desarrollado por DeepMind de Google, lo resolvió con una precisión asombrosa. Fue una revolución, un triunfo de la ingeniería computacional que prometía acelerar el descubrimiento de fármacos y desvelar los secretos más profundos de la biología.
AlphaFold2 era como un maestro arquitecto, un sistema que había estudiado durante años los planos evolutivos de la vida, utilizando un conocimiento biológico profundo para construir una maquinaria predictiva de una complejidad abrumadora. Su método se basaba en una idea brillante: analizar los «Alineamientos de Secuencias Múltiples» (MSA, por sus siglas en inglés), una técnica que compara la misma proteína a través de millones de especies para encontrar pistas evolutivas sobre su estructura. Su arquitectura interna era una obra de arte de la especialización, con módulos diseñados a medida para interpretar esta información.
Ahora, apenas unos años después de esa hazaña histórica, emerge un nuevo protagonista que propone una filosofía radicalmente distinta. Su nombre es SimpleFold, y es el resultado de una investigación llevada a cabo por un equipo de científicos de Apple. Si AlphaFold2 fue el maestro arquitecto que construyó su obra maestra con herramientas especializadas y un profundo conocimiento del dominio, SimpleFold es el aprendiz brillante que logra resultados comparables con un conjunto de herramientas mucho más simple y universal.
Este nuevo sistema abandona deliberadamente la complejidad de su predecesor. No necesita los laboriosos alineamientos de secuencias evolutivas ni los módulos de red neuronal diseñados a medida. En su lugar, se apoya en la elegancia y la versatilidad de la arquitectura Transformer, la misma tecnología que impulsa a los grandes modelos de lenguaje como GPT. SimpleFold trata el plegamiento de proteínas como un problema de generación, similar a cómo una IA crea una imagen a partir de una descripción de texto. Utiliza una técnica llamada flow-matching para «esculpir» la estructura tridimensional de una proteína a partir de una nube aleatoria de átomos, guiada únicamente por la secuencia de aminoácidos.
Este artículo se adentra en el corazón de esta segunda revolución. Exploraremos por qué el plegamiento de proteínas es un pilar fundamental de la vida y una fuente de enfermedad. Desentrañaremos la genialidad y la complejidad de AlphaFold2 para comprender el paradigma que estableció. Y, finalmente, analizaremos en profundidad la audaz simplicidad de SimpleFold, un sistema que no solo desafía las convenciones, sino que también sugiere un nuevo y emocionante futuro para la inteligencia artificial en la ciencia: un futuro donde el poder no reside en la complejidad artesanal, sino en la escala y la generalidad del aprendizaje.
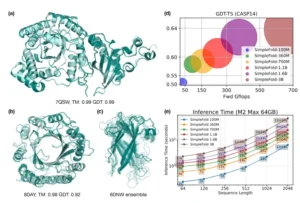
Aquí se ven ejemplos de las predicciones de SimpleFold para las proteínas en (a) y (b), donde la forma real se ve en aguamarina claro y el resultado de la inteligencia artificial en verde azulado intenso. El panel (c) muestra un grupo de posibles estructuras generadas para una proteína, demostrando cómo el modelo es capaz de capturar su flexibilidad y movimiento natural. El gráfico (d) ilustra cómo la precisión general de SimpleFold aumenta notablemente conforme las versiones del modelo se hacen más grandes y potentes. El gráfico final (e) mide el tiempo que tardan las distintas versiones de SimpleFold en un ordenador potente de uso normal, como lo es un MacBook Pro de 64 GB.
El código secreto de la vida y el enigma de su forma
Para comprender la magnitud del desafío que sistemas como SimpleFold y AlphaFold2 han abordado, primero debemos sumergirnos en el mundo de la biología molecular y apreciar el papel central que desempeñan las proteínas. Si el ADN es el libro de recetas de la vida, que contiene las instrucciones para construir un organismo, las proteínas son los chefs, los obreros y los mensajeros que ejecutan esas instrucciones. Son las moléculas que hacen casi todo el trabajo en las células y son esenciales para la estructura, función y regulación de los tejidos y órganos del cuerpo.
Todo comienza con una simple cadena. Las proteínas se construyen a partir de un alfabeto de solo veinte aminoácidos diferentes, unidos uno tras otro en una secuencia lineal, como las cuentas de un collar. La longitud de esta cadena puede variar desde unas pocas docenas hasta miles de aminoácidos. Sin embargo, en esta forma lineal, la proteína es inerte, una simple hebra sin propósito. Para volverse funcional, debe someterse a un proceso de plegamiento espontáneo, un acto de autoorganización que la transforma en una estructura tridimensional compleja y altamente específica. Esta forma final, o «conformación nativa», está determinada enteramente por la secuencia de aminoácidos y las leyes de la física y la química que gobiernan sus interacciones. Fuerzas como los enlaces de hidrógeno, las interacciones hidrofóbicas y las atracciones electrostáticas guían la cadena para que se tuerza y se pliegue en hélices, láminas y bucles, formando una arquitectura molecular única.
La precisión de este plegamiento es de una importancia crítica. La función de una proteína depende de su forma, de la misma manera que la función de una llave depende de la forma de sus dientes. Una enzima, por ejemplo, tiene un «sitio activo» con una geometría precisa que le permite unirse a una molécula específica y catalizar una reacción química. Un anticuerpo tiene una forma que le permite reconocer y neutralizar a un invasor patógeno. Si el plegamiento falla, la función se pierde.
Peor aún, un plegamiento incorrecto puede tener consecuencias catastróficas. Cuando las proteínas no adoptan su forma correcta, pueden volverse «pegajosas» y comenzar a agruparse, formando agregados tóxicos que interfieren con la función celular normal. Este fenómeno de mal plegamiento y agregación es la causa subyacente de una clase de dolencias devastadoras conocidas como proteinopatías. En la enfermedad de Alzheimer, la proteína beta-amiloide se pliega incorrectamente y se acumula en placas que dañan las neuronas.

En la enfermedad de Parkinson, la proteína alfa-sinucleína forma agregados llamados cuerpos de Lewy, que también son tóxicos para las células cerebrales. La enfermedad de Huntington es causada por una mutación que produce una versión anormalmente larga de la proteína huntingtina, que se pliega mal y se agrega, mientras que en la fibrosis quística, una mutación en el gen CFTR provoca que la proteína resultante se pliegue incorrectamente y sea destruida por la célula antes de que pueda llegar a la membrana celular para realizar su función de transporte de iones.
Esta conexión directa entre la forma de una proteína y la salud humana es lo que convierte el problema del plegamiento en una cuestión de urgencia médica. Si los científicos pudieran predecir con fiabilidad la estructura 3D de cualquier proteína a partir de su secuencia de aminoácidos, se abrirían puertas sin precedentes para el diseño de fármacos. Sería posible diseñar moléculas que se ajusten perfectamente a los sitios activos de las enzimas de un patógeno para desactivarlas, o crear fármacos que estabilicen una proteína propensa al mal plegamiento, o incluso diseñar terapias que bloqueen la formación de agregados tóxicos. Durante décadas, determinar la estructura de una proteína fue un proceso arduo, costoso y, a menudo, infructuoso, que dependía de técnicas experimentales como la cristalografía de rayos X. La promesa de resolver este problema computacionalmente representaba nada menos que un santo grial para la biología y la medicina.
La primera revolución: cuando la IA descifró el enigma
Cada dos años desde 1994, la comunidad de biólogos computacionales se reúne para un evento singular: la «Evaluación Crítica de Técnicas para la Predicción de la Estructura de Proteínas», o CASP. Más que una simple conferencia, CASP es el campeonato mundial de esta disciplina, un riguroso experimento a doble ciego diseñado para medir objetivamente el estado del arte en la predicción de estructuras proteicas. Los organizadores distribuyen secuencias de aminoácidos de proteínas cuyas estructuras acaban de ser resueltas experimentalmente pero aún no se han hecho públicas. Equipos de todo el mundo envían sus predicciones computacionales, que luego se comparan con las estructuras reales. Es el campo de pruebas definitivo, donde las nuevas ideas se validan o se descartan.
Durante años, el progreso fue lento e incremental. Los métodos mejoraban, pero ninguna técnica lograba consistentemente la precisión necesaria para ser verdaderamente útil en aplicaciones del mundo real. Todo cambió en CASP14, en el año 2020. Un participante, AlphaFold2 de DeepMind, logró un rendimiento tan extraordinario que dejó atónita a la comunidad científica. Sus predicciones alcanzaron una precisión comparable a la de los métodos experimentales, resolviendo de manera efectiva un gran desafío científico que había perdurado durante 50 años. Fue un momento decisivo, una revolución que demostró el poder de la inteligencia artificial para resolver problemas fundamentales en la ciencia.
La clave del éxito sin precedentes de AlphaFold2 residía en su ingenioso uso de la información evolutiva. El sistema no intentaba resolver el problema únicamente a partir de las leyes de la física. En cambio, realizaba un profundo trabajo de «detective evolutivo». Su primer paso era tomar la secuencia de la proteína objetivo y buscar en gigantescas bases de datos genómicas para encontrar miles de secuencias similares en otras especies, desde bacterias hasta humanos. Este conjunto de secuencias relacionadas se organizaba en lo que se conoce como un Alineamiento de Secuencias Múltiples (MSA).
La lógica detrás del MSA es brillante: si dos aminoácidos en una proteína están muy alejados en la secuencia lineal pero han mutado de forma coordinada a lo largo de millones de años de evolución (es decir, cuando uno cambia, el otro también lo hace para compensar), es muy probable que estén en contacto físico en la estructura 3D plegada. Al analizar estos patrones de coevolución en un MSA profundo, AlphaFold2 podía inferir una red de restricciones espaciales, un mapa de qué aminoácidos debían estar cerca unos de otros.
Para procesar esta vasta cantidad de información relacional, DeepMind diseñó una arquitectura de red neuronal altamente especializada y computacionalmente intensiva. El sistema mantenía dos representaciones de la información en paralelo: una para la secuencia de aminoácidos y otra, llamada «representación de pares» (pair representation), para las relaciones entre cada par de aminoácidos.
El corazón de AlphaFold2 era un módulo llamado «Evoformer», que contenía mecanismos de atención complejos, incluidas las «actualizaciones triangulares» (triangle updates), diseñados específicamente para refinar iterativamente estas dos representaciones, permitiendo que la información fluyera de un lado a otro. Era una maquinaria intrincada, una obra maestra de la ingeniería de IA que codificaba explícitamente el conocimiento biológico sobre la evolución en su diseño.
Sin embargo, esta dependencia del MSA, la fuente misma de su poder, también representaba una vulnerabilidad. Para las llamadas «proteínas huérfanas», aquellas que tienen pocos o ningún pariente evolutivo conocido, el MSA es superficial o inexistente, y el rendimiento de AlphaFold2 puede disminuir. Además, el proceso de buscar y construir un MSA profundo es un cuello de botella computacional, que requiere una cantidad significativa de tiempo y recursos antes de que la predicción pueda comenzar. Este compromiso entre precisión y dependencia evolutiva definió el paradigma de la primera revolución y, sin saberlo, abrió la puerta a una nueva filosofía que buscaría alcanzar una meta similar a través de un camino mucho más directo y general.

Una nueva filosofía: la elegancia de la simplicidad
El advenimiento de SimpleFold no representa un intento de destronar a AlphaFold2, sino de proponer una ruta alternativa, una que se basa en principios de generalidad y escalabilidad en lugar de especialización y conocimiento de dominio codificado. La filosofía central de SimpleFold es un cambio de perspectiva: en lugar de tratar el plegamiento de proteínas como un problema de inferencia estructural a partir de datos evolutivos, lo aborda como una tarea de generación condicional.
La analogía más poderosa y accesible para entender este enfoque proviene del mundo de la IA generativa de imágenes. Modelos como DALL-E o Midjourney han cautivado al mundo con su capacidad para crear imágenes fotorrealistas a partir de una simple descripción de texto. El usuario proporciona un «prompt», como «un astronauta montando a caballo en la luna al estilo de Van Gogh», y el modelo genera una imagen que corresponde a esa descripción. SimpleFold opera bajo un principio conceptualmente idéntico. La secuencia de aminoácidos de una proteína actúa como el «prompt», y el modelo genera el resultado deseado: la estructura tridimensional completa de todos sus átomos en el espacio.
Para lograr esta hazaña, SimpleFold abandona la arquitectura a medida de AlphaFold2 y adopta en su lugar una de las herramientas más versátiles y potentes de la inteligencia artificial moderna: la arquitectura Transformer. Presentada por primera vez en 2017 en el influyente artículo «Attention is All You Need», la arquitectura Transformer revolucionó el procesamiento del lenguaje natural y se convirtió en la base de todos los grandes modelos de lenguaje actuales. Su poder no reside en módulos especializados, sino en un mecanismo fundamental llamado «autoatención» (self-attention).
La autoatención puede describirse como la capacidad de un modelo para sopesar la importancia de todas las demás partes de una secuencia de entrada al procesar una parte específica. En el contexto del lenguaje, esto permite que un modelo entienda que en la frase «el robot cogió la pelota y la lanzó», la palabra «la» se refiere a «pelota», aunque estén separadas por otras palabras. SimpleFold aplica este mismo principio a la secuencia de aminoácidos. El mecanismo de autoatención le permite evaluar las relaciones entre todos los aminoácidos de la cadena simultáneamente. Aprende de forma dinámica qué residuos, sin importar cuán distantes estén en la secuencia lineal, necesitan interactuar y estar cerca en la estructura 3D.
Este enfoque generalista le permite a SimpleFold prescindir de los componentes especializados que eran cruciales para AlphaFold2. No necesita una «representación de pares» explícita ni las complejas «actualizaciones triangulares», porque el mecanismo de autoatención del Transformer puede aprender estas relaciones espaciales directamente de los datos. En lugar de depender de un MSA, SimpleFold utiliza un modelo de lenguaje de proteínas (PLM) preentrenado, en este caso ESM2, para convertir la secuencia de aminoácidos en una rica representación numérica que ya captura información implícita sobre la biología de la proteína. La arquitectura Transformer actúa como una navaja suiza de la IA, una herramienta de propósito general cuya eficacia proviene de su capacidad para aprender patrones complejos en cualquier tipo de datos secuenciales, ya sean palabras en una oración, píxeles en una imagen o, como en este caso, aminoácidos en una proteína.
Esculpiendo proteínas a partir del ruido: la magia del flow-matching
El motor que impulsa la capacidad generativa de SimpleFold es una técnica matemática sofisticada pero conceptualmente elegante conocida como flow-matching. Para apreciar su innovación, es útil contrastarla con el enfoque generativo más conocido en la actualidad: los modelos de difusión.
Los modelos de difusión, que sustentan a muchos de los generadores de imágenes más populares, funcionan a través de un proceso de dos pasos: uno de destrucción y otro de reconstrucción. Imagine que tiene una fotografía perfectamente nítida (la estructura proteica final y correcta).
El «proceso hacia adelante» de la difusión consiste en añadir gradualmente una pequeña cantidad de ruido gaussiano (estática aleatoria) a la imagen en cientos o miles de pasos. Al final de este proceso, la imagen original se ha transformado en un patrón de ruido puro e irreconocible. El objetivo del entrenamiento es enseñar a una red neuronal a revertir este proceso. El modelo aprende a predecir y eliminar el ruido en cada paso, comenzando con una imagen de pura estática y «desenrrollando» el proceso de difusión paso a paso hasta reconstruir una imagen clara y coherente. Es un método poderoso, pero inherentemente iterativo y, a veces, lento.
El flow-matching, o ajuste de flujos, ofrece un camino más directo. La analogía aquí no es la de restaurar una foto borrosa, sino la de un escultor que trabaja con un bloque de arcilla. El proceso comienza con una distribución simple y conocida, como una nube de átomos dispersos aleatoriamente en el espacio (el bloque de arcilla). El objetivo es transformar esta nube inicial en la distribución compleja y específica que representa la estructura proteica final (la estatua terminada).
En lugar de aprender a revertir un proceso de adición de ruido, un modelo de flow-matching aprende un «campo de velocidad» o un «flujo». Este campo de velocidad es como un conjunto de instrucciones increíblemente detalladas que le dice a cada átomo en la nube exactamente en qué dirección y a qué velocidad debe moverse en cada instante de tiempo para llegar a su posición final correcta.
El entrenamiento consiste en mostrarle al modelo el punto de partida (ruido) y el punto final (la estructura proteica real) y enseñarle a predecir el camino en línea recta que los conecta. El modelo aprende a generar un flujo continuo que transporta suavemente los átomos desde un estado de desorden a uno de orden perfecto. La ventaja de este enfoque es su simplicidad y eficiencia.
El objetivo de entrenamiento es más directo, a menudo una simple regresión de mínimos cuadrados para que el campo de velocidad predicho coincida con el campo de velocidad real. Además, una vez entrenado, el proceso de generación puede ser más rápido, ya que el modelo ha aprendido un camino directo y no necesita deshacer un proceso de difusión paso a paso.
SimpleFold aplica esta técnica para generar la estructura completa de una proteína, incluyendo todos los átomos de la cadena principal y las cadenas laterales. El modelo se entrena con un objetivo de pérdida combinado. La parte principal es la pérdida de flow-matching, que se define como:
donde el modelo vθ aprende a predecir la velocidad correcta para pasar de una estructura ruidosa xt a la estructura final x. Además, se incluye un término de pérdida estructural auxiliar, el Test de Diferencia de Distancia Local (LDDT), que mide directamente el error en las distancias entre pares de átomos en la estructura generada. Esta pérdida adicional ayuda al modelo a refinar los detalles locales de la geometría atómica, asegurando que los enlaces y ángulos sean físicamente realistas. Esta combinación de un objetivo generativo elegante y una restricción estructural directa es la clave de la capacidad de SimpleFold para producir estructuras de alta calidad.
SimpleFold en acción: rendimiento, escala y eficiencia
Una nueva filosofía arquitectónica y un elegante mecanismo generativo son prometedores en teoría, pero su verdadero valor se mide en el campo de batalla de los benchmarks científicos. El equipo detrás de SimpleFold evaluó su familia de modelos en dos de las pruebas más rigurosas y respetadas en el campo: CAMEO22, una evaluación continua y automatizada, y el legendario CASP14, el mismo certamen donde AlphaFold2 hizo historia.
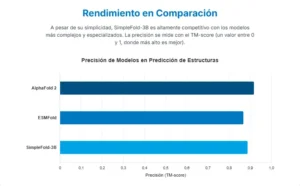
Los resultados demuestran que la simplicidad no implica un sacrificio de rendimiento. SimpleFold-3B, el modelo más grande con 3 mil millones de parámetros, logra un rendimiento «competitivo» en comparación con los modelos de vanguardia más complejos. En el benchmark CAMEO22, se acerca notablemente a los resultados de gigantes como AlphaFold2 y RoseTTAFold2, y en el más desafiante CASP14, supera a ESMFold, otro modelo de alto rendimiento que también prescinde de los MSA. El verdadero triunfo no es necesariamente superar a estos modelos en todas las métricas, sino alcanzar un nivel de precisión comparable eliminando por completo los costosos módulos de diseño específico como las actualizaciones triangulares y la búsqueda de MSA.
La siguiente tabla resume el rendimiento de SimpleFold frente a otros modelos clave, ilustrando esta competitividad. Los valores se presentan como media/mediana sobre todas las muestras de prueba.
Comparativa de rendimiento en plegamiento de proteínas (CAMEO22 y CASP14)
| Tipo | Modelo | TM-score ↑ | GDT-TS ↑ | LDDT ↑ | LDDT-Ca ↑ | RMSD ↓ |
| CAMEO22 | ||||||
| MSA-basado | AlphaFold2 | 0.863/0.942 | 0.844/0.903 | 0.845/0.904 | 0.893/0.923 | 3.578/1.857 |
| MSA-basado | RoseTTAFold2 | 0.864/0.947 | 0.816/0.856 | 0.727/0.767 | 0.893/0.926 | 3.571/1.707 |
| PLM-basado | ESMFold | 0.853/0.933 | 0.826/0.875 | 0.792/0.834 | 0.867/0.906 | 3.973/2.019 |
| PLM-basado | SimpleFold-100M | 0.803/0.878 | 0.746/0.787 | 0.721/0.752 | 0.822/0.844 | 4.897/2.855 |
| PLM-basado | SimpleFold-360M | 0.826/0.905 | 0.782/0.841 | 0.773/0.803 | 0.852/0.878 | 4.775/2.681 |
| PLM-basado | SimpleFold-700M | 0.829/0.915 | 0.788/0.845 | 0.775/0.809 | 0.850/0.886 | 4.557/2.423 |
| PLM-basado | SimpleFold-1.1B | 0.833/0.924 | 0.793/0.851 | 0.776/0.807 | 0.850/0.883 | 4.350/2.334 |
| PLM-basado | SimpleFold-1.6B | 0.835/0.916 | 0.799/0.864 | 0.782/0.816 | 0.853/0.889 | 4.397/2.187 |
| PLM-basado | SimpleFold-3B | 0.837/0.916 | 0.802/0.867 | 0.773/0.802 | 0.852/0.884 | 4.225/2.175 |
| CASP14 | ||||||
| MSA-basado | AlphaFold2 | 0.845/0.907 | 0.778/0.817 | 0.783/0.855 | 0.856/0.897 | 5.027/3.015 |
| PLM-basado | ESMFold | 0.701/0.792 | 0.622/0.711 | 0.637/0.705 | 0.715/0.802 | 8.679/4.016 |
| PLM-basado | SimpleFold-100M | 0.611/0.628 | 0.513/0.544 | 0.549/0.537 | 0.685/0.659 | 11.157/8.976 |
| PLM-basado | SimpleFold-360M | 0.674/0.758 | 0.617/0.657 | 0.585/0.654 | 0.703/0.762 | 9.382/4.828 |
| PLM-basad | SimpleFold-700M | 0.680/0.767 | 0.630/0.674 | 0.591/0.668 | 0.714/0.763 | 9.289/4.431 |
| PLM-basado | SimpleFold-1.1B | 0.697/0.796 | 0.640/0.676 | 0.607/0.668 | 0.723/0.758 | 9.249/4.462 |
| PLM-basado | SimpleFold-1.6B | 0.712/0.801 | 0.630/0.709 | 0.660/0.699 | 0.741/0.798 | 8.424/4.722 |
| PLM-basado | SimpleFold-3B | 0.720/0.792 | 0.639/0.703 | 0.666/0.709 | 0.747/0.829 | 7.732/3.923 |
Sin embargo, donde SimpleFold realmente brilla y demuestra la superioridad de su enfoque generativo es en la generación de ensambles. Las proteínas no son estructuras estáticas y rígidas; son moléculas dinámicas que respiran, se flexionan y cambian de forma para llevar a cabo sus funciones. Los modelos entrenados con objetivos de regresión determinista, como AlphaFold2 o ESMFold, están diseñados para predecir una única estructura de baja energía. SimpleFold, al ser un modelo generativo, puede modelar la distribución completa de posibles conformaciones. Al introducir un grado controlado de aleatoriedad en el proceso de generación, puede producir no una, sino un conjunto o «ensamble» de estructuras plausibles que una proteína podría adoptar. Esta capacidad es de un valor incalculable para el diseño de fármacos.
A menudo, un fármaco se une a una conformación transitoria de una proteína, en un «bolsillo críptico» que solo es visible cuando la proteína se flexiona de una manera particular. Al generar un ensamble de estructuras, SimpleFold puede revelar estas oportunidades ocultas para la intervención terapéutica, una hazaña que es intrínsecamente difícil para los modelos no generativos.
El trabajo también presenta una evidencia convincente sobre la escalabilidad del enfoque. Los autores entrenaron una familia de modelos SimpleFold, desde uno pequeño de 100 millones de parámetros hasta el gigante de 3 mil millones, y demostraron una clara correlación: a mayor tamaño del modelo y mayor cantidad de datos de entrenamiento (hasta casi 9 millones de estructuras destiladas), mejor es el rendimiento, especialmente en los benchmarks más difíciles. Esto sugiere que la arquitectura generalista de SimpleFold aún no ha alcanzado su techo de rendimiento y que futuros aumentos en la escala computacional y la disponibilidad de datos podrían llevar a mejoras aún mayores.
Finalmente, uno de los aspectos más sorprendentes y prácticos de SimpleFold es su eficiencia. A pesar de su tamaño, la arquitectura basada en Transformers estándar es computacionalmente más eficiente que los complejos módulos de AlphaFold2. Esto se traduce en tiempos de inferencia notablemente rápidos. El documento muestra que incluso el modelo de 3 mil millones de parámetros puede predecir la estructura de una proteína de longitud media en menos de un minuto en hardware de consumo, como un MacBook Pro con un chip M2 Max. Esta eficiencia representa un paso significativo hacia la democratización de estas poderosas herramientas, poniéndolas al alcance de un número mucho mayor de investigadores en todo el mundo, sin necesidad de acceso a supercomputadoras.

El futuro del diseño biológico: más allá del plegamiento
La presentación de SimpleFold es mucho más que el anuncio de un nuevo modelo de predicción de proteínas. Es el heraldo de un cambio de paradigma en la forma en que la inteligencia artificial aborda los problemas científicos complejos. Representa una transición desde un enfoque de «conocimiento codificado» hacia uno de «aprendizaje emergente». La primera revolución, liderada por AlphaFold2, demostró que se podía resolver un gran desafío científico diseñando intrincadamente una IA para que pensara como un biólogo evolutivo. Esta segunda revolución, iniciada por SimpleFold, sugiere que una arquitectura de aprendizaje lo suficientemente general y potente, alimentada con una cantidad masiva de datos, puede aprender por sí misma las reglas fundamentales de la física y la biología que gobiernan el plegamiento de proteínas, sin necesidad de que se le enseñen explícitamente.
Esta distinción tiene implicaciones profundas. Al reducir la dependencia de módulos de red neuronal altamente especializados y heurísticas de dominio, este nuevo enfoque reduce la barrera de entrada para la innovación. Los investigadores de otros campos científicos, desde la ciencia de los materiales hasta la climatología, pueden ahora ver un camino más claro para aplicar estas poderosas arquitecturas de propósito general a sus propios desafíos, sin necesidad de convertirse en expertos en el diseño de redes neuronales a medida. La lección de SimpleFold es que la escalabilidad de la arquitectura y los datos puede ser una estrategia más fructífera a largo plazo que la especialización artesanal.
El futuro que esta nueva filosofía desbloquea es uno en el que la IA pasa de ser una herramienta para predecir la naturaleza a convertirse en un socio para diseñarla. La arquitectura simplificada y adaptable de SimpleFold es inherentemente más flexible. Se puede ajustar y extender con mayor facilidad para abordar desafíos relacionados pero distintos que van más allá de la predicción de estructuras nativas.
Podemos vislumbrar un futuro cercano en el que modelos basados en estos principios se utilicen para el diseño de novo de proteínas: la creación de proteínas completamente nuevas con funciones que no existen en la naturaleza. Esto podría incluir enzimas diseñadas para descomponer plásticos en el medio ambiente, catalizadores biológicos para procesos industriales más eficientes y sostenibles, o proteínas con propiedades terapéuticas novedosas. En el campo de la vacunología, estos modelos podrían acelerar el diseño de inmunógenos altamente estables y eficaces contra futuros patógenos. En la inmunoterapia, podrían permitir la ingeniería de «biológicos» a medida, como anticuerpos o receptores de células T, diseñados para atacar células cancerosas con una especificidad sin precedentes.
SimpleFold, por lo tanto, no es el final de la historia, sino el comienzo de un nuevo y emocionante capítulo. Al demostrar que el plegamiento de proteínas es, en efecto, más simple de lo que pensábamos, ha abierto un vasto espacio de diseño para el progreso futuro. Es una invitación a la comunidad científica para construir modelos generativos más eficientes, potentes y, sobre todo, más generales, que puedan aprender los principios fundamentales del mundo natural directamente a partir de los datos. Es un paso crucial en el camino hacia una nueva era de descubrimiento molecular, una en la que la sinergia entre la inteligencia humana y la artificial nos permitirá no solo comprender el código de la vida, sino también reescribirlo para el bien de la humanidad.
Referencias
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ronneberger, O., Willmore, L., Ballard, A. J., Bambrick, J., Bodenstein, S. W., Evans, D. A., Hung, C.-C., O’Neill, M., Reiman, D., Tunyasuvunakool, K., Wu, Z., Zemgulyte, A., Arvaniti, E., … Jumper, J. M. (2024). Accurate structure prediction of biomolecular interactions with alphafold 3. Nature, 630, 493–500.
Ahdritz, G., Bouatta, N., Floristean, C., Kadyan, S., Xia, Q., Gerecke, W., O’Donnell, T. J., Berenberg, D., Fisk, I., Zanichelli, N., et al. (2024). Openfold: Retraining alphafold2 yields new insights into its learning mechanisms and capacity for generalization. Nature methods, 21(8), 1514–1524.
Albergo, M. S., Boffi, N. M., & Vanden-Eijnden, E. (2023). Stochastic interpolants: A unifying framework for flows and diffusions. arXiv preprint arXiv:2303.08797.
Albergo, M. S., & Vanden-Eijnden, E. (2023). Building normalizing flows with stochastic interpolants. En The Eleventh International Conference on Learning Representations (ICLR).
Peebles, W., & Xie, S. (2023). Scalable diffusion models with transformers. En ICCV.
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., Kaiser, Ł., & Polosukhin, I. (2017). Attention is all you need. Advances in neural information processing systems, 30.
Wang, Y., Lu, J., Jaitly, N., Susskind, J., & Bautista, M. A. (2025). SimpleFold: Folding Proteins is Simpler than You Think. arXiv preprint arXiv:2509.18480.

